

李红良教授团队发现病理性心肌肥厚调控新机制
2022年4月19日,李红良教授团队在Circulation Research(IF=17)在线发表题为“The E3 Ligase TRIM16 Is a Key Suppressor of Pathological Cardiac Hypertrophy”的研究论文,该研究首次揭示了TRIM16是病理性心肌肥厚的关键保护分子,通过靶向Prdx1并抑制其磷酸化,抑制病理性心肌肥厚,有望成为改善病理性心肌肥厚的治疗靶点。


图片源自Circulation Research
当心脏长期处于病理性应激状态下,将会诱发病理性心肌肥厚,主要表现为心肌细胞体积增大、间质和血管周围纤维化、蛋白合成增多和肌成纤维细胞活化,如果这一系列心室重构改变不能在短时间内得到改善,将会逐步恶化并最终导致心力衰竭和猝死。心肌肥厚的发病机制极其复杂,已有研究表明,机械拉伸、氧化应激和炎症等在病理性心肌肥厚的发生发展中具有重要的作用。尽管已经进行了大量研究,但尚未批准特效药物干预心肌肥厚。因此,发现心肌肥厚的特异性调控靶点将为其提供新的治疗策略。
本研究基于临床相关性探索与转录组学数据库分析,发现TRIM16在病理性心肌肥厚以及心力衰竭样本中受EGR2转录调控而应激性上调;并与样本心肌肥厚及心力衰竭的程度呈显著正相关。团队通过转录组测序结果发现,TRIM16抑制心肌肥厚、氧化应激、蛋白合成等分子事件的关键基因表达且TRIM16过表达和敲低对关键事件的调控作用完全相反。通过构建心肌细胞特异性TRIM16基因修饰小鼠,团队发现心肌细胞特异性敲除TRIM16明显加重病理性心肌肥厚及纤维化,并且导致心功能下降,而过表达 TRIM16可 有效改善心肌结构和心功能。
分子研究及验证实验阐明,TRIM16与Prdx1相互作用并抑制其磷酸化,增强其下游Nrf2-HO-1通路,从而改善氧化应激,抑制心肌细胞肥大。深入研究发现TRIM16调控Prdx1磷酸化的具体机制;TRIM16依赖于其E3泛素连接酶活性,通过促进Src K48泛素化降解从而抑制Prdx1磷酸化。
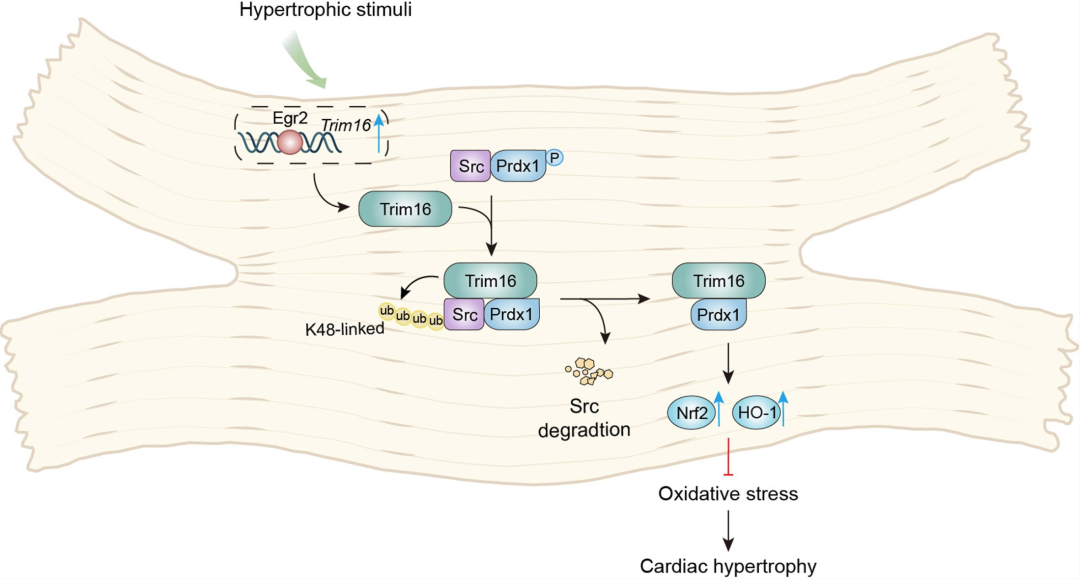
文章模式图图源自Circulation Research
本研究揭示了TRIM16蛋白调控的新机制,提供了TRIM16抗心肌肥厚的首个证据,为病理性心肌肥厚的临床治疗提供了潜在的分子靶点与新的方向。
过去十四年来,李红良教授团队 专注于 心血管疾病与代谢性疾病领域 , 探索这些疾病的基本发病机制并开发治疗策略。团队的研究成果在世界知名期刊 Nature Medicine, Cell Metabolism, Sci Transl Med,Circulation, Circulation Research, Nature Communications,PNAS, Hepatology, 等上发表研究论文累计 200 余篇 。
为您推荐
算法反馈精品有声
热门文章
精彩视频


凤凰资讯官方微信




好书精选



